Über Alpha-Mannosidose
Alpha-Mannosidose im Überblick
Was ist Alpha-Mannosidose?
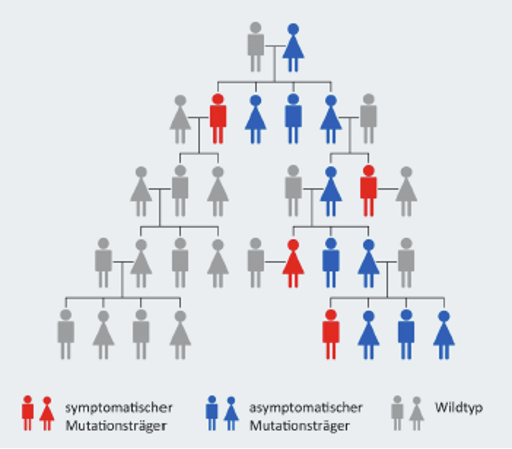
Alpha-Mannosidose ist eine genetisch bedingte Erkrankung aus der Gruppe der sogenannten lysosomalen Speichererkrankungen. Sie gehört zu den seltenen Erkrankungen und tritt bei ca. 1-2 von 1.000.000 Menschen auf. Verursacht wird sie durch verschiedene Veränderungen, sogenannten Mutationen, in den Abschnitten des Erbmaterials, die für die Ausprägung des lysosomalen Enzyms Alpha-Mannosidase verantwortlich sind.
Früher wurde die Alpha-Mannosidose in drei Typen unterteilt: leichte Form, mittelschwere Form und schwere Form. Da die einzelnen Formen fließend ineinander übergehen können, ist diese strenge Einteilung inzwischen allerdings nicht mehr üblich.
Das klinische Bild der Alpha-Mannosidose ist insgesamt individuell sehr variabel. Es handelt sich aber stets um eine progressive, also voranschreitende Erkrankung mit großem Einfluss auf die Lebensqualität, da im Krankheitsverlauf meist das Skelett, das Gehör, das Nervensystem und das Immunsystem der Patient*innen betroffen sind.3
Informativer Kurzfilm zur Alpha-Mannosidose
In diesem kurzen Film finden Sie umfassende Informationen zur Diagnostik, zu den wichtigsten Symptomen und zur Therapie der Alpha-Mannosidose. Frau Prof. Dr. Julia B. Hennermann, die sich auf pädiatrische Stoffwechselerkrankungen spezialisiert hat und Prof. Dr. Dag Malm, der maßgeblich zur Erforschung dieser lysosomalen Speichererkrankungen beigetragen hat und selbst Vater von zwei erkrankten Töchtern ist, vermitteln Ihnen hier ein umfassendes Bild.
Ursachen und Vererbungsmuster
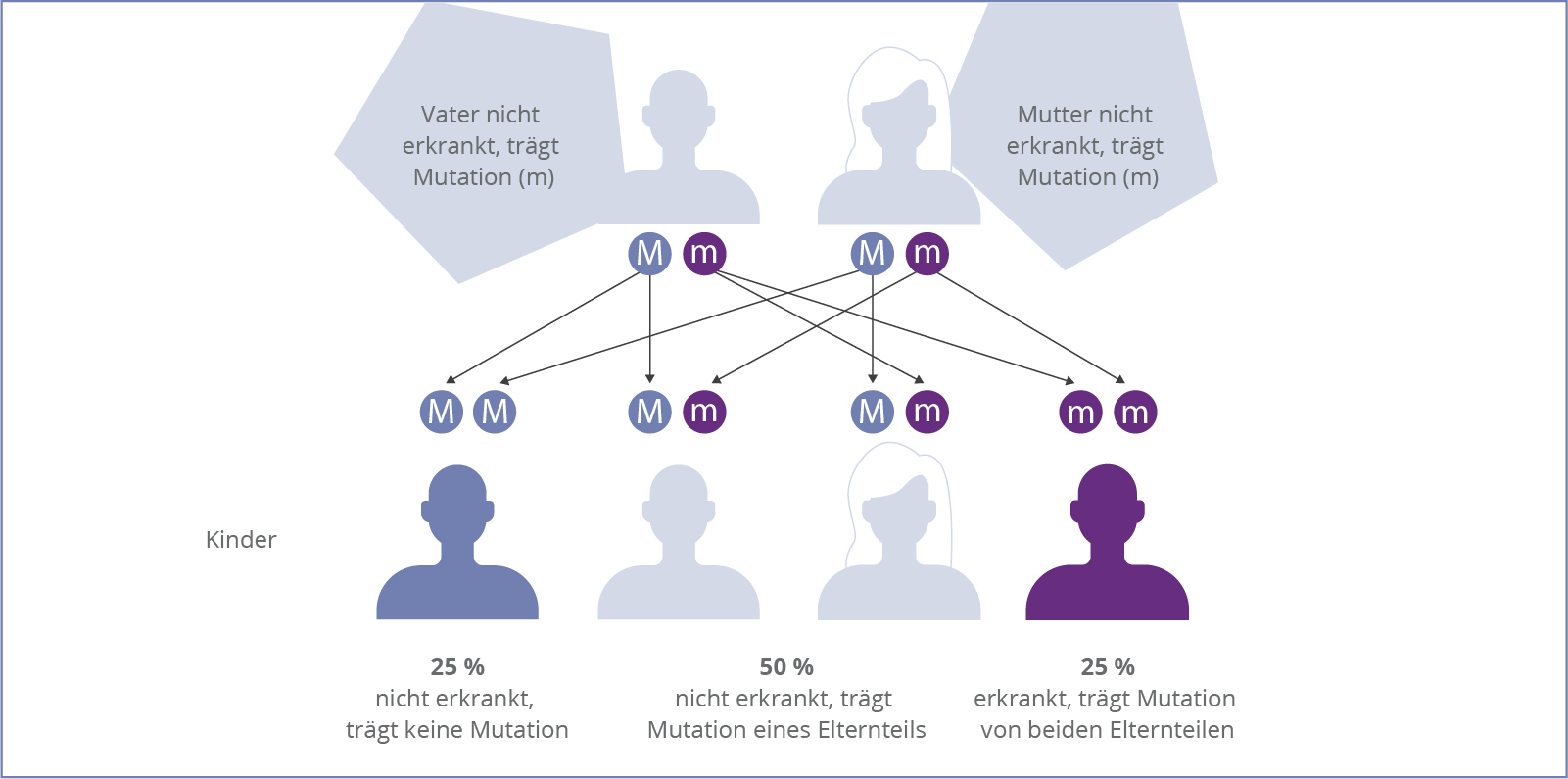
Die Alpha-Mannosidose ist eine autosomal-rezessiv vererbte Krankheit, d.h. die Erkrankung tritt nur dann in Erscheinung, wenn sich auf den jeweiligen Chromosomen beider Elternteile eine krankmachende Veränderung befindet. Durch diesen Defekt wird der Abbau von mannosehaltigen Mehrfachzuckern gestört, sogenannter Oligosacchariden.
Da es eine Vielzahl möglicher Mutationen gibt, ist es für ein Paar, das bereits ein Kind mit Alpha-Mannosidose hat, ratsam, sich bezüglich der weiteren Familienplanung an eine genetische Beratung zu wenden. So kann ein besseres Verständnis der Betroffenen gegenüber der Erkrankung geschaffen werden.9
Was passiert bei Alpha-Mannosidose im Körper?
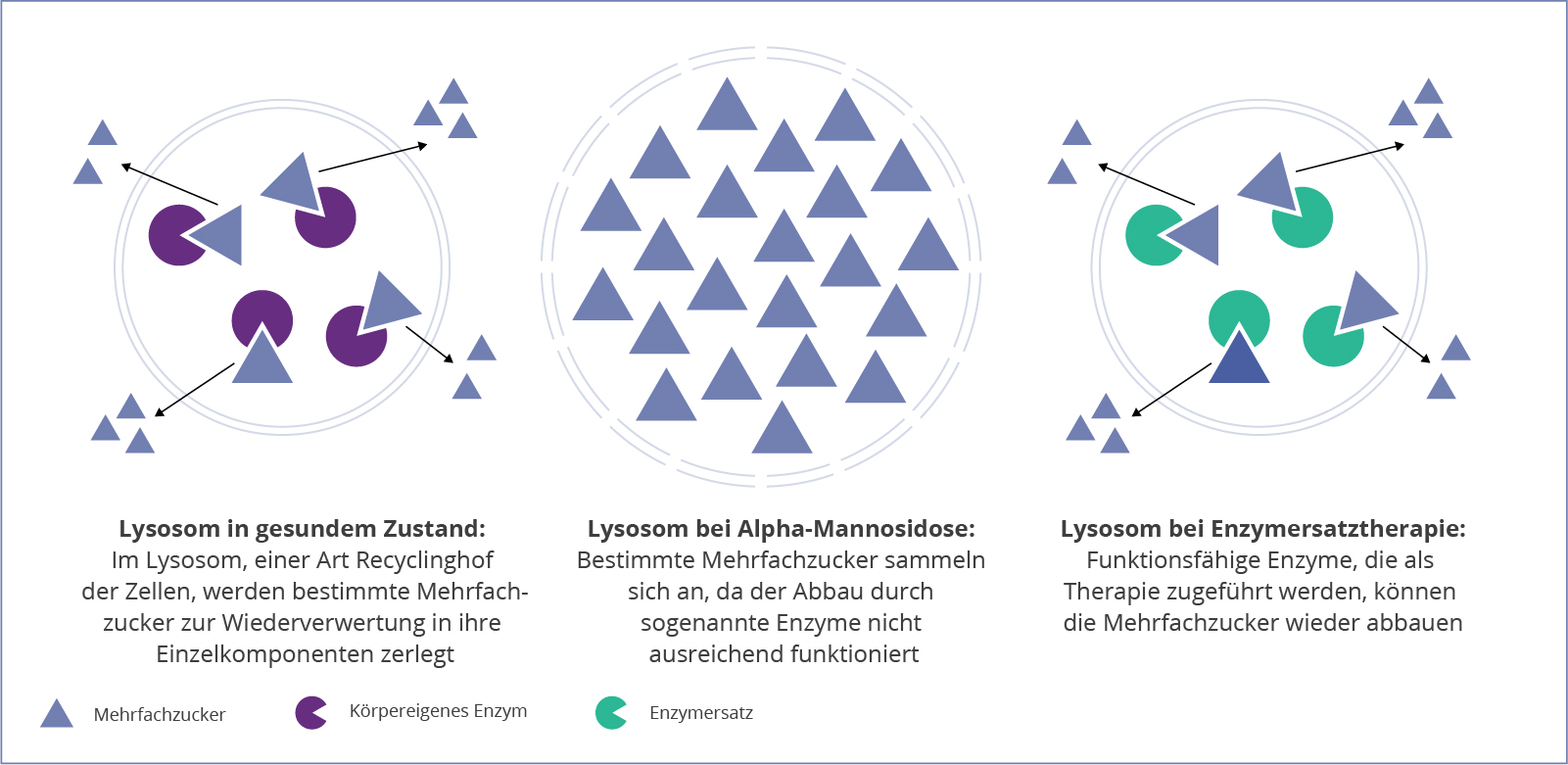
Als lysosomale Speichererkrankung steht die Alpha-Mannosidose mit der Ansammlung einer Zuckerart, der Mannose, in den Zellen in Zusammenhang. Die Erkrankung nimmt dabei ihren Anfang in den Lysosomen, den “Recyclinghöfen” der Zellen. Durch einen Defekt im Gen MAN2B1, wird ein bestimmtes Enzym nicht erfolgreich gebildet. Diese Glykosidase ist normalerweise für den Abbau von hybriden und komplexen mannosereichen Oligosacchariden im Lysosom verantwortlich.1,3
Der Auf- und Abbau der Oligosaccharide ist ein entscheidender Mechanismus für verschiedene Bereiche des Körpers. Diese komplexen Zucker sind unter anderem beim Aufbau von Knochen, Knorpel, Sehnen oder Haut beteiligt. Dabei findet in den Lysosomen ein ständiges Recycling dieser Moleküle statt, wobei neue Oligosaccharide produziert und alte abgebaut werden. Durch einen Funktionsverlust oder das Fehlen der Alpha-Mannosidase, sammeln sich die Oligosaccharide mit der Zeit in den Lysosomen an, was zur Schädigung und letztlich zum Absterben der Zellen führen kann.1,3
Verlauf und Therapie der Alpha-Mannosidose
Das klinische Bild der Alpha-Mannosidose kann sich im Krankheitsverlauf ändern. Unter Verlauf und Prognose finden Sie mehr Informationen über die fortschreitende Erkrankung in den verschiedenen Lebensphasen. Für die Alpha-Mannosidose stehen inzwischen Therapiemöglichkeiten zur Verfügung, die von einer ursächlichen Enzymersatztherapie über eine symptomatische Behandlung bis hin zur Unterstützung durch Hilfsmittel wie etwa Gehhilfen reichen.2,10 Möchten Sie mehr zur Therapie der Alpha-Mannosidose erfahren, folgen Sie diesem Link.
- Beck M, et al. Orphanet J Rare Dis. 2013;8:88.
- Malm D, Nilssen Ø. Orphanet J Rare Dis. 2008;3:21.
- Borgwardt L et al. Orphanet J Rare Dis. 2015;10:70.
- Neufeld EF, et al. The Metabolic and Molecular Bases of Inherited Disease. 2001;3421–3452.
- Grosse H. Pädiatrie. 2018;30,30.
- Beck, M. Dtsch Arztebl. 2001;98:34-35.
- Alpha-Mannosidosis Mutation Database. Tromsoe University. https://apex.jupiter.no/apex/f?p=101:1 (zuletzt aufgerufen: April 2022).
- Ceccarini MR, et al. Int J Mol Sci. 2018;19:1500.
- Guide to understanding mannosidosis. Society for Mucopolysaccharide Diseases. http://www.mpssociety.org.uk/wp-content/uploads/2016/07/guide-alphamannosidosis-2013.pdf (zuletzt aufgerufen: April 2022).
- Fachinformation Lamzede®
Symptome
So äußert sich Alpha-Mannosidose
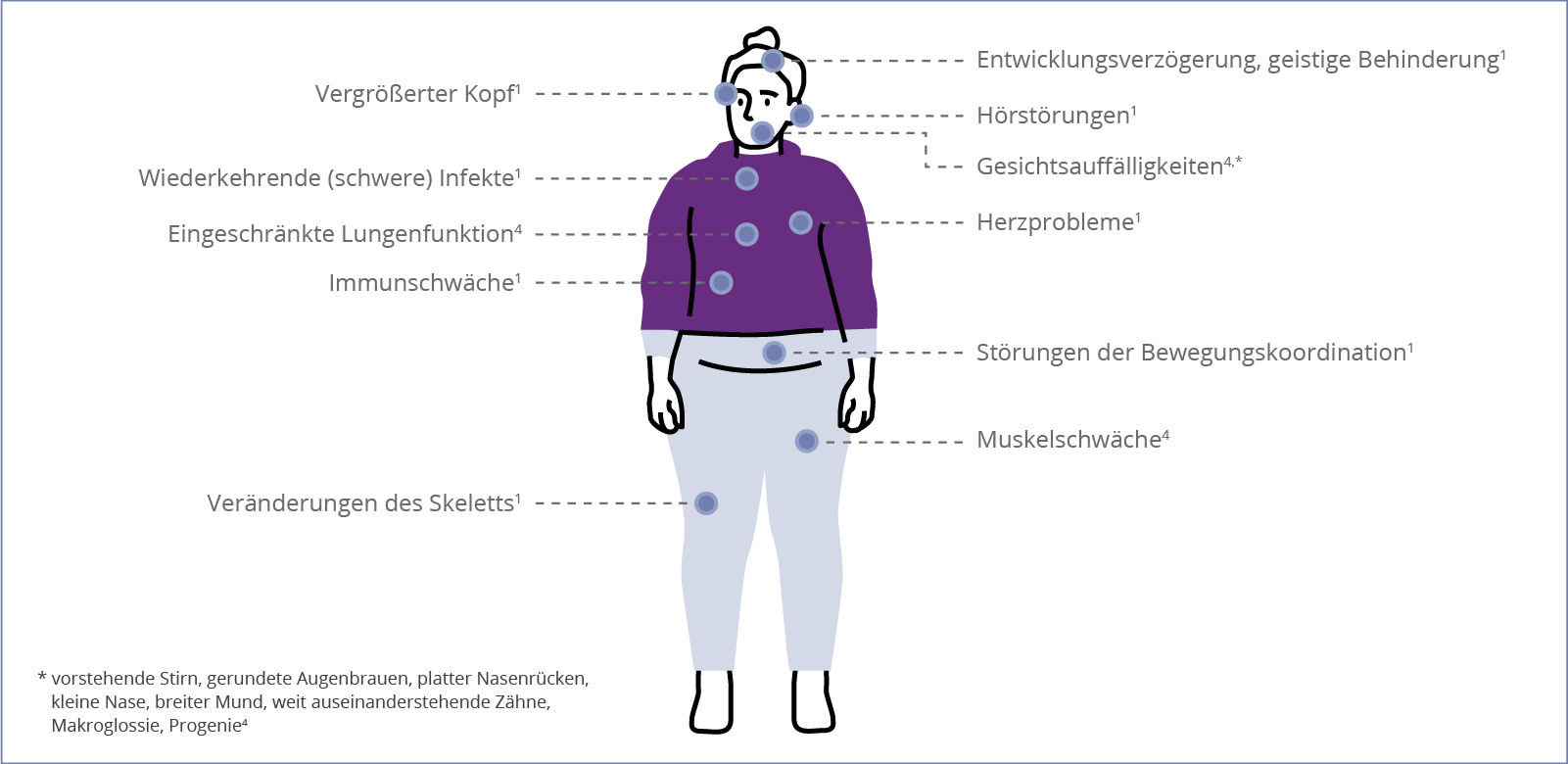
Die möglichen Auswirkungen der Alpha-Mannosidose auf den Körper von Betroffenen sind sehr variabel. Da es sich um eine progressive, also voranschreitende, Erkrankung handelt, kann sich zusätzlich die Ausprägung der Beschwerden im Laufe der Zeit verändern. Durch die Vielzahl an möglichen Symptomen und die verschiedenen, individuellen Krankheitsverläufe, dauert es oft sehr lange, bis die richtige Diagnose gestellt wird.1 Hier finden Sie eine Übersicht der typischen physischen und psychischen Krankheitsmerkmale:
Gesichtszüge
Es gibt verschiedene Anzeichen im Gesicht von Betroffenen, die auf eine Alpha-Mannosidose hinweisen können. Typische Auffälligkeiten im Gesicht sind:1,3
- Übernormal großer Schädel (Makrozephalie)
- Stark ausgeprägte Stirn
- Abgerundete Augenbrauen
- Abgeflachter Nasenrücken
- Lückenstand der Zähne
- Vorstehender Oberkiefer (Prognathie)
- Vergrößerte Zunge (Makroglossie)
- Verkürzung des Nackens
- Hydrocephalus (bei sehr schwerer Ausprägung der Alpha-Mannosidose)
Gehör
Die meisten Betroffenen erleiden ab der frühen Kindheit eine Beeinträchtigung ihres Gehörs. Dabei tritt sowohl konduktiver Hörverlust (Schallleitungsschwerhörigkeit) als auch sensorineuraler Hörverlust (Schallempfindungsschwerhörigkeit) auf. Entzündungen des Gehörgangs und Paukenergüsse können hier das Krankheitsbild verschlimmern. Unbehandelt führt der Hörverlust in der frühen Kindheit meist zu Störungen des Sprachvermögens und der geistigen Fähigkeiten.1,3
Augen
Weitere sensorische Beeinträchtigungen im Zuge der Alpha-Mannosidose betreffen häufig die Augen der Patient*innen. Symptome wie Weitsichtigkeit (Hyperopie), Kurzsichtigkeit (Myopie) und Schielen (Strabismus) sind dabei weit verbreitet. Selten treten auch Veränderungen der Linsen und eine oberflächliche Trübung der Hornhaut auf.3
Immunsystem
Vor allem in der Kindheit erleiden Betroffene häufig schwere, wiederkehrende Infektionen, da die Immunantwort des Körpers gestört ist. Durch die Alpha-Mannosidose besteht eine verminderte Fähigkeit spezifische Antikörper zu bilden. Außerdem zeigen die Leukozyten (weiße Blutkörperchen) eine reduzierte Aktivität auf, wodurch schwere Krankheitsverläufe bei bakteriellen Infektionen entstehen.1,3
Skelett
- Die Alpha-Mannosidose wirkt sich auf unterschiedliche Weise auf das Skelett der Betroffenen aus. Folgende Symptome können dabei auftreten:1,3
- Asymptomatische Osteopenie
- Fokale oder sklerotische Läsionen
- Osteonekrose
- Dysostosis multiplex (bei 90 % der Patient*innen)
- Verdickung der Schädeldecke (Calvaria)
- Deformierte Wirbelkörper
- Hypoplasie
- Erweiterung der Röhrenknochen in den Händen
- Deformierungen des Brustbeins (Sternum) und der Wirbelsäule
- X-Beine (Genu valgum)
- Erkrankung mehrerer Gelenke (Polyarthropathie - vor allem Hüftgelenksarthrose und Kniearthrose)
Bewegung
Die häufigste Störung der Bewegungskoordination im Zuge einer Alpha-Mannosidose ist die Ataxie (Bewegungsstörung). Die Entwicklung motorischer Fähigkeiten ist allgemein verlangsamt und betroffene Kinder werden als tollpatschig wahrgenommen. Verursacher dieser Störungen können eine Muskelschwäche, Gelenkprobleme oder Ataxien in Folge einer Hirnatrophie (Gehirnschwund) oder demyelinisierenden Erkrankung (Entmarkungskrankheit) sein. Neben Anomalien der Gelenke, Myopathie (Muskelschwäche) und Muskelhypotonie (verminderte Muskelgrundspannung) führt die Krankheit auch zu Störungen in Gehirnbereichen, die für motorische Fähigkeiten und Muskelkoordination zuständig sind. In seltenen Fällen erleiden die Betroffenen spastische Paralysen. Die Auswirkungen auf den Bewegungsapparat der Patient*innen verlaufen meist fortschreitend.1,3
Psyche
- Geistige Behinderung1,3
Die frühe Entwicklung der Psychomotorik von Patient*innen mit Alpha-Mannosidose scheint meist normal zu verlaufen, doch in fast allen Fällen besteht eine geistige Behinderung. Im Krankheitsverlauf kann sich eine zunächst leichte geistige Behinderung (IQ 60-80) weiter verschlechtern. Dabei nehmen sowohl die Sprachfunktion als auch geistige und motorische Fähigkeiten im Laufe der Zeit immer weiter ab. Die gestörte Sprachentwicklung hängt oft mit einem Hörverlust zusammen und macht sich durch ein beschränktes Vokabular und verminderte Verständlichkeit bemerkbar. - Psychiatrische Symptome1,3
Etwa 25 % der Betroffenen leiden unter psychiatrischen Problemen, welche meist im Laufe der Pubertät eintreten. Das Spektrum reicht dabei von Verwirrtheitszuständen über Angststörungen und Depressionen bis hin zu Halluzinationen. Psychosen und anschließende Hypersomnien (Schlafsucht) können ebenso auftreten. Die psychiatrischen Symptome werden häufig durch einen Verlust von kognitiven Fähigkeiten wie das Sprechen oder Lesen begleitet.
- Malm D, Nilssen Ø. Orphanet J Rare Dis. 2008;3:21.
- Beck M, et al. Orphanet J Rare Dis. 2013;8:88.
- Malm D, Nilssen Ø. Alpha-Mannosidosis. NCBI. 2001. https://www.ncbi.nlm.nih.gov/books/NBK1396/ (Zuletzt aufgerufen: April 2022)
- Grosse H. Pädiatrie. 2018;30:30.
Diagnose
Diagnoseverfahren & Differenzialdiagnosen
Alpha-Mannosidose kann mit verschiedenen Tests diagnostiziert werden, die sowohl auf biochemischen als auch auf molekulargenetischen Verfahren basieren. Eine sogenannte Trockenblutkarte ermöglicht eine Kombination dieser Tests und sichert als Stufendiagnostik eine sichere Diagnose.
Differenzialdiagnostik
Das klinische Erscheinungsbild der Alpha-Mannosidose ist dem anderer lysosomaler Speicherkrankheiten sehr ähnlich, wie z.B. den Mukopolysaccharidosen (MPS).1 Daher erfolgt bei Verdacht auf MPS auch immer eine Differenzialdiagnose zur Alpha-Mannosidose – und umgekehrt. Die Trockenbluttests anhand einer Trockenblutkarte ermöglichen hier schnell und einfach eine solche Differenzialdiagnose, da hier sowohl die Enzymaktivität der MPS, als auch die der Alpha-Mannosidose untersucht wird und ein vorliegender Enzymdefekt rasch erkannt werden kann.
Vorgehen
Zur eindeutigen Diagnosestellung der Alpha-Mannosidose werden verschiedene Faktoren beachtet. Dazu gehören: die Identifizierung der charakteristischen Symptome, die Analyse der Krankenakte und die Ergebnisse verschiedener diagnostischer Tests. Diese umfassen sowohl enzymatische Analysen, als auch molekulargenetische Untersuchungen.
Diagnose-Algorithmen
Eine Gruppe führender internationaler Expert*innen hat für den Ablauf einer sicheren und schnellen Diagnosestellung zwei Diagnose-Algorithmen entwickelt. Bei jüngeren Betroffenen (Patient*innen unter 10 Jahren) gelten Hörschädigungen und Sprachprobleme als Hauptsymptome, die zum Verdacht einer Alpha-Mannosidose führen. Werden keine weiteren Symptome gefunden, können die Patient*innen entweder unter weitere Beobachtung gestellt oder an ein Stoffwechselzentrum überwiesen werden. Hier erfolgen weitere Tests wie die Bestimmung der Oligosaccharid-Konzentration im Urin und/oder enzymatische Analysen. Molekulargenetische Tests können dann eine Diagnose final bestätigen.3
Bei älteren Patient*innen (älter als 10 Jahre) gelten geistige Behinderungen, koordinative Störungen und/oder psychische Auffälligkeiten als Hauptsymptome einer Alpha-Mannosidose. Liegen diese Anzeichen vor, sollte eine klinische Bewertung der Krankenakte erfolgen, um herauszufinden, ob sich in der Vergangenheit bereits Symptome bemerkbar gemacht haben. Trifft dies nicht zu, können weitere Beobachtungen und Tests oder eine Überweisung an ein Stoffwechsel-Zentrum erfolgen.3
- Malm D, Nilssen Ø. Orphanet J Rare Dis. 2008;3:21.
- Alpha-Mannosidose, Diagnostik. Amedes. https://www.di-ch.de/diagnostik.html, zuletzt aufgerufen: Mai 2022.
- Guffon N, et al. Mol Gen Metab. 2019;126:470.
Verlauf und Prognose
Krankheitsverlauf
Das klinische Erscheinungsbild der Alpha-Mannosidose besteht aus einer Reihe von leichten bis schwerwiegenden Symptomen. Daher hat die Erkrankung auch je nach Ausprägung einen anderen Verlauf.
Verlauf
Die Alpha-Mannosidose verläuft progressiv, d.h. die Symptome steigern sich allmählich mit zunehmendem Lebensalter. Bei erkrankten Neugeborenen treten zunächst meist keine Symptome auf, was die Diagnose verzögern kann. Im Laufe des Lebens wirkt sich die multisystemische Erkrankung dann u. a. auf die Muskulatur und das Skelett der Betroffenen aus. Es kann zu starken Beeinträchtigungen im Alltag kommen, was die Lebensqualität senkt. Ausdauer, Beweglichkeit, Atmung und die neurokognitive Entwicklung nehmen häufig immer weiter ab. Schwer erkrankte Patient*innen können letzten Endes dann auf Hilfsmittel wie einen Rollstuhl oder dauerhafte Hilfe durch andere angewiesen sein. Es kann zu sozialer Isolation, Verhaltensstörungen und psychiatrischen Problemen kommen.1,2
Insbesondere wenn die Alpha-Mannosidose nicht behandelt wird, können durch den progressiven Verlauf in den verschiedenen Altersgruppen folgende Symptome auftreten:
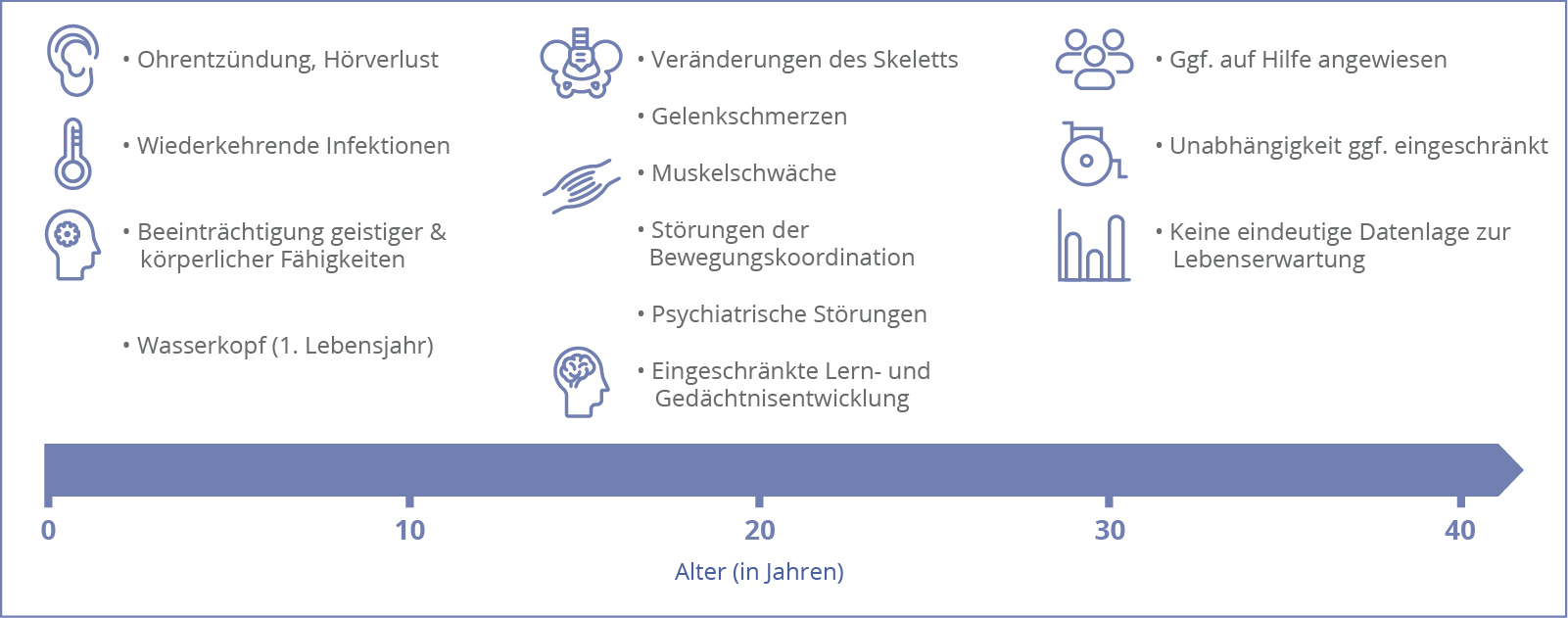
Klicken Sie hier, um mehr Informationen über die Symptome bei einer Alpha-Mannosidose zu erhalten.
Der Zeitpunkt des Auftretens der Symptome steht mit der Ausprägung der Erkrankung in Zusammenhang. Es wurde beobachtet, dass eine schwere Form in der Kindheit häufig zu einem sehr schweren Verlauf und einem frühen Versterben der Betroffenen führt.1,3 Folgende Übersicht verschiedener Ausprägungsformen kann als Orientierung dienen, allerdings ist diese strenge Einteilung nicht mehr üblich, da die einzelnen Formen fließend ineinander übergehen und individuell sehr variabel sein können.
Prognose
Es gibt verschiedene Therapiemöglichkeiten, um eine Alpha-Mannosidose zu behandeln und so ihren Verlauf mildern zu können. Die Folgen der Skelettanomalien können mit symptomatischen Behandlungen, wie orthopädischen Operationen oder konservativen Therapien, eingeschränkt werden. Kausale Therapieoptionen wie eine hämatopoetische Stammzelltransplantation können bei der Erhaltung der neurokognitiven Funktion helfen und ein Fortschreiten der Krankheit verhindern.1 Außerdem können Auswirkungen der Erkrankung auf den psychischen Zustand Betroffener beispielsweise durch einen Wechsler-Test (Intelligenztest) beobachtet werden. Durch eine Enzymersatztherapie kann das immunologische Profil Betroffener verbessert werden. Diese Therapieform hatte in bisherigen Studien auch positive Effekte auf motorische Funktionen und die Lungenfunktion der Patient*innen.4-6 Die Behandlung der Alpha-Mannosidose ist sehr komplex und umfasst verschiedene medizinische Gebiete. Daher ist es notwendig, ein breit aufgestelltes Expertenteam für die Therapie zu Rate zu ziehen, um den Gesundheitszustand Betroffener verbessern zu können.
- Malm D, Nilssen Ø. Orphanet J Rare Dis. 2008;3:21.
- Borgwardt L et al. Pediatr Endocrinol Rev. 2014;12 Suppl 1:185-91.
- Alpha Mannosidosis. National Organization for Rare Diseases (NORD) Factsheet 2015. https://rarediseases.org/rare-diseases/alpha-mannosidosis/, zuletzt aufgerufen: Mai 2022.
- Borgwardt et al. J Inherit Metab Dis. 2018.
- Lund AM. et al. J Inherit Metab Dis. 2018.
- Harmatz P. et al. Mol Genet Metab. 2018.
Therapie & Nebenwirkungen
Therapiemanagement bei Alpha-Mannosidose
Die Therapie der Alpha-Mannosidose besteht sowohl aus der Behandlung der verschiedenen Krankheitssymptome als auch aus kausalen Therapieansätzen. Hier wird versucht die Ursache der Krankheit zu beseitigen. Durch das multisystemische Erkrankungsbild müssen dabei viele verschiedene medizinische Bereiche in die Behandlung miteinbezogen werden.
Symptomatische Behandlung
Um Patient*innen mit Alpha-Mannosidose vollumfänglich zu therapieren ist ein Expertenteam aus Bereichen wie der Otolaryngologie (Hals-Nasen-Ohren-Heilkunde), Logopädie oder Orthopädie nötig. In folgender Tabelle ist eine Übersicht möglicher Therapieansätze in den verschiedenen medizinischen Gebieten dargestellt.1
| Infektiologie | Otolaryngologie | Logopädie | Orthopädie |
|---|---|---|---|
|
|
|
|
Außerdem sollten präventive Maßnahmen eingeleitet werden, um eine weitere Verschlechterung des Gesundheitszustandes Betroffener zu verhindern. Dazu gehören neben prophylaktischen Impfungen auch regelmäßige körperliche Untersuchungen, die Kontrolle des Wachstums und neurophysiologische Tests. Dabei wird stets dokumentiert, ob Depressionen, Fatigue (Müdigkeit & Erschöpfung), Infektionen, Gewichtsverlust, Schmerzen, Bewegungseinschränkungen oder andere Änderungen des Gesundheitszustandes bei den Patient*innen auftreten.2
Kausale Therapieansätze
Bisher stehen zwei verschiedene Therapiemöglichkeiten zur Verfügung, um die Alpha-Mannosidose spezifisch zu behandeln:
- Hämatopoetische Stammzelltransplantation (HSZT)
Bei der HSZT werden gesunde Spenderzellen, die das Enzym Alpha-Mannosidase sekretieren können, in das Empfängergewebe transplantiert. Diese Therapieoption wird vor allem bei sehr jungen Patient*innen unter 3 Jahren empfohlen, da hier die Erkrankung des Nervensystems noch nicht allzu stark fortgeschritten ist. Durch eine HSZT können neurokognitive Funktionen erhalten und ein Fortschreiten der Krankheit verhindert werden.1-3
Eine HSZT geht allerdings mit gewissen Risiken einher. So steigt beispielsweise die Wahrscheinlichkeit, eine sogenannte Graft-versus-Host-Reaktion (eine Abwehrreaktion) zu erleiden. Es hat sich gezeigt, dass vor allem bei älteren Patient*innen Komplikationen auftraten. Der Nutzen des Verfahrens sollte vor einer Behandlung stets mit den Risiken abgewogen werden.1-3
- Enzymersatztherapie (EET)
Durch einen Enzymersatz wird der fehlende lysosomale Abbau mannosereicher Oligosaccharide korrigiert. Das defekte Enzym Alpha-Mannosidase wird dabei durch ein biotechnologisch hergestelltes Enzym ersetzt und die Ansammlung der Oligosaccharide (Mehrfachzucker/Kohlenhydrate) in den Lysosomen somit reduziert. Durch die EET können die motorische Funktion, die Lungenfunktion und das immunologische Profil Betroffener verbessert werden. Da bei der EET die Blut-Hirn-Schranke nicht überwunden wird, ist nicht davon auszugehen, dass Auswirkungen auf neurologische Manifestationen oder eine Beeinträchtigung kognitiver Funktionen auftreten.1,4-8 Häufig auftretende Nebenwirkungen umfassen moderate Symptome wie Gewichtszunahme, Diarrhoe oder Kopfschmerzen.
- Malm D, Nilssen Ø. Alpha-mannosidosis. Orphanet J Rare Dis 2008;3:21.
- Malm D, Nilssen Ø. Alpha-Mannosidosis. NCBI. 2019. https://www.ncbi.nlm.nih.gov/books/NBK1396/ (Zuletzt aufgerufen: Mai 2022)
- Grewal SS et al. J Pediatr 2004;144:569-573.
- Borgwardt et al. J Inherit Metab Dis. 2018.
- Lund AM. et al. J Inherit Metab Dis. 2018.
- Harmatz P. et al. Mol Genet Metab. 2018.
- Ardigò D. et al. J Inborn Err Metab Screening; 2016.
- Guffon N, Konstantopoulou V, Hennermann JB, et al. J Inherit Metab Dis. 2023